
Research | DOI: https://doi.org/10.31579/2640-1053/090
*Corresponding Author: Erlinda M. Gordon, Cancer Center of Southern California, Santa Monica CA 90403, US.
Citation: J R. Ravicz, CW. Szeto, S Reddy, S P. Chawla, M A. Morse, F L. Hall, E M. Gordon. (2021) CCNG1 oncogene: a novel biomarker for cancer therapy /gene therapy. J.Cancer Research and Cellular Therapeutics. 5(4); Doi: 10.31579/2640-1053/090
Copyright: © 2021 Erlinda M. Gordon, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 09 July 2021 | Accepted: 21 July 2021 | Published: 03 August 2021
Keywords: deltarex-g; human cyclin g1; cell cycle control; cancer gene therapy; oncogenic drivers; cancer stem cells; cell competence
Background: Metastatic cancer is associated with an invariably fatal outcome. However, DeltaRex-G, a tumor-targeted retrovector encoding a gene-edited dominant-negative CCNG1 inhibitor gene, has induced long term (>10 years) survival of patients with chemo-resistant metastatic pancreatic adenocarcinoma, malignant peripheral nerve sheath tumor, osteosarcoma, B-cell lymphoma, and breast carcinoma.
Objective: To evaluate the level of CCNG1 expression in tumors as a potential biomarker for CCNG1 (Cyclin G1-blocking) inhibitor therapy.
Methods: CCNG1 RNA expression levels that were previously measured as part of whole genome molecular profiling of tumors (TCGA, N=9161), adjacent “tissues” (TCGA, N=678) and GTEx normal tissues (N=7187) across 22 organ sites were analyzed. Differential expression of CCNG1 and Ki-67 in primary (N= 9161) vs metastatic (N= 393) tumors were also compared in primary (N=103) vs. metastatic (N=367) skin cancers (i.e., melanoma).
Statistical Analysis: To detect systematically differential expression of CCNG1 and Ki-67 expression between populations (e.g. tumor vs. normal), unpaired Student's t-tests were performed.
Results: Enhanced CCNG1 RNA and Cyclin G1 protein expression were noted in tumors compared to normal analogous counterparts, and CCNG1 expression correlated significantly with that of Ki-67. Moreover, CCNG1 expression tended to be higher than that of Ki-67 in metastatic vs primary tumors.
Conclusions: Taken together with the emerging Cyclin G1 / Cdk / Myc / Mdm2 / p53 Axis governing Cancer Stem Cell Competence, this supportive data indicates: (1) CCNG1 expression is frequently enhanced in tumors when compared to their normal analogous counterparts, (2) CCNG1 and Ki-67 expressions are higher in metastatic vs primary tumors, (3) CCNG1 expression is significantly correlated with that of Ki-67, and (4) CCNG1 may actually be a stronger prognostic marker of stem cell competence, chemo-refractoriness, and EMT/metastasis than Ki-67. Phase 2 studies are planned to identify patients most likely to respond favorably to CCNG1 inhibitor therapy.
Cyclins are a highly conserved family of executive regulatory proteins that govern the molecular-genetic “activation” of quiescent stem cells (i.e., Cell Competence to proliferate), as well as the definitive phases of cell cycle progression, in both normal tissue regeneration and in disease, through physical interaction with cognate cyclin-dependent proline-directed protein kinase partners (CDKs) [1,2], which phosphorylate and regulate key regulatory substrates exerting cell cycle checkpoint control. The human Cyclin G proto-oncogene (CCNG1) was originally identified/cloned, silenced-experimentally, blocked-functionally (by gene-edited dominant-negative mutation), and characterized physiologically in the crucible of clinical oncology: thereby defining “The pivotal Cyclin G1 / Cdk / Myc / Mdm2 / p53 Axis” as a prospective Unifying Theme in oncology [3,4]—identifying Cyclin G1 expression profile as a frequent feature and key oncogenic driver of cancer stem cell survival [5], c-Myc oncogenicity [6], loss of DNA fidelity [7], cancer development [8], progression [9], epithelial-to-mesenchymal transition (EMT) [9], refractoriness to chemotherapies [10], and the troublesome immune anergy [11] of advanced metastatic cancers.
The CCNG1 oncogene-a non-canonical cyclin exhibiting molecular aspects of both transcriptional regulation and cell cycle control [1,2] operates at the “first and rate-limiting step” in the animal cell cycle: that is, stem cell Competence (G0 to G1 boundary), where Cyclin G1/CDK activity phosphorylates and stabilizes the c-Myc oncogene/transcription factor [12-14], contributing to cancer progression and metastatic behavior [3-6]; while the physical association of Cyclin G1 with the Mdm2 oncogene, a ubiquitin ligase, is a negative regulator of the TP53 (p53) tumor suppressor: a guardian of DNA fidelity [7], which is commonly lost with cancer progression. As such, CCNG1 has been clinically determined to be a strategic target for cancer gene therapy.
DeltaRex-G—previously developed clinically as Rexin-G [15-19]—is a tumor-targeted amphotropic MLV-based retroviral vector: (i) displaying a collagenous Pathotropic Signature (SIG)-binding/targeting peptide on its gene-edited gp70 envelope protein [20,21], and (ii) encoding a gene-edited dominant-negative expression construct of the CCNG1 oncogene (dnG1)—the enforced expression of dnG1 blocks Cyclin G1-dependent cell survival function(s) and enforces apoptosis in a broad spectrum of cancer cells in vitro, in animal models, and clinically in advanced metastatic cancers [3,4] where notable safety, efficacy and long-term survival have been achieved (> 12 years) with repeated infusions of DeltaRex-G [22-24]. When administered intravenously, the tumor-targeted nanoparticles (~100 nm) actively seek-out and accumulate-in the tumor microenvironment (TME), binding with high-affinity to abnormal (anaplastic) SIG proteins exposed biochemically during tumor invasion, tumor angiogenesis, and/or reactive stroma formation [25]. Treatment with DeltaRex-G has demonstrated objective clinical benefits and prolonged survival in metastatic, chemotherapy-resistant pancreatic ductal adenocarcinoma, osteosarcoma, sporadic malignant peripheral nerve sheath tumor (sMPNST), and both hormone receptor positive and triple negative ductal carcinoma of breast [3,4, 22-24]. In light of the pivotal roles and survival function of Cyclin G1 in cancer stem cell biology [5-11], the observed clinical efficacy of DeltaRex-G, including long-term survival benefits achieved in advanced, metastatic, chemo-resistant cancers, suggests that DeltaRex-G may indeed be most efficacious and comparatively advantageous in advanced metastatic tumors that overexpress the pivotal and commanding CCNG1 oncogene. Here, the aims of this investigation are to report augmented CCNG1 oncogene expression in many solid tumors and provide the rational basis for profiling Cyclin G1expression in tumors to identify patients who are likely to benefit from CCNG1 inhibitor therapy.
All archived tumor samples are de-identified tissue samples previously used for other histopathologic examination and molecular profiling studies at NantOmics Bioinformatics Department of Nant and at the Cancer Center of Southern California/Sarcoma Oncology Research Center. Written consent was obtained from patients as part of histopathologic examination and molecular profiling of archived tumors as standard of care.
The Genotype-Tissue [removed]GTEx) project is an ongoing effort to build a comprehensive public resource to study tissue-specific gene expression and regulation. Samples were collected from 54 non-diseased tissue sites across nearly 1000 individuals, primarily for molecular assays including WGS, WES, and RNA-Seq. Remaining samples are available from the GTExBio bank. The GTEx Portal provides open access to data including gene expression, QTLs, and histology images.
CCNG1 RNA Sequence Analysis
Previously obtained whole-transcriptome expression levels across 22 commensurate organ sites within three different settings: tumor tissues (The Cancer Genome Atlas [TCGA], n=9161), tumor-adjacent tissues (TCGA, n=678) and normal tissues (Genotype-Tissue Expression Portal [GTEx], n=7187) were analyzed for CCNG1 expression.
Qualification of IHC assay for Cyclin G1 expression
An anti-cyclin G1 IgG1 kappa light chain murine monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, cat# sc-8016), coupled with MACH4 anti-mouse secondary antibody (BioCare Medical, Pacheco, CA) were used for immunohistochemical (IHC) analysis. The mouse monoclonal antibody was optimized on cell line tissue microarray and normal tissue microarray samples sectioned at approximately 4 μm and adhered to positively charged glass. Multiple antigen retrieval methods and a series of titrations of the cyclin G1 antibody were performed using the tissue microarray slides to determine the optimal antigen retrieval method and antibody concentration. Once the antibody concentration was chosen, accuracy, specificity, sensitivity, and the range of staining intensity was assessed using de-identified samples of hepatocellular carcinoma and breast carcinoma samples as positive controls, and normal tissues as negative controls. Additionally, three neoplastic tissues representing various degrees of cyclin G1 expression were chosen for an assessment of reproducibility and precision. Then, IHC analysis was performed on 27 de-identified formalin-fixed, deparaffinized samples, representing 13 types of cancer and 4 types of normal tissue. The optimized method is as follows: First, the samples were deparaffinized and rehydrated, and endogenous peroxidase activity was blocked. Then, antigen retrieval was performed in a decloaking chamber with Tris-EDTA for 20 minutes at 95° C. The anti-cyclin G1 antibody was applied at a dilution of 1:2000, and incubated for 30 minutes at room temperature, followed by application of the MACH4 mouse secondary antibody for 15 minutes at room temperature. A MACH4 HRP Polymer (BioCare Medical, Pacheco, CA) was then applied as the detection system, and incubated for 30 minutes at room temperature. The resulting product was incubated in chromogen DAB-beta (BioCare Medical, Pacheco, CA) for 10 minutes at room temperature, and was then counterstained with hematoxylin, dehydrated, cleared in xylene, and finally a coverslip was applied. The samples were then analyzed by a board-certified pathologist who scored the staining intensities of the samples on a scale of 0 to 3+.
Reproducibility was tested in several ways: (1) intra-run reproducibility, with identical slides run in triplicate in one run on one day; (2) inter-run reproducibility with identical slides tested on three separate runs in three separate days; (3) inter-tech reproducibility, with identical slides run by two different technologists in two separate runs; and (4) inter-instrument reproducibility with identical slides run by the same technologist on two different instruments. Concordance factors were calculated based on the pathologist’s scoring of the reproducibility slides. As an indirect confirmation of cyclin G1 assay specificity, additional IHC experiments were performed to compare expression of cyclin G1 to that of cellular proliferation marker Ki-67. Intra-run, inter-run, inter-tech, and inter-instrument reliability were all deemed as acceptable and CCNG1 expression observed by IHC was in line with that predicted in the literature.
To generate statistically comparable expression profiles for each sample, transcripts-per-million (TPM) values were generated using RSEM [26] scaled using only protein-coding transcripts, then converted to (approximately) Gaussian distributions by log2 transformation (i.e. log2[TPM+1]).
To detect systematically differential expression of CCNG1 and Ki-67 expression between populations (e.g. tumor vs. normal), unpaired Student's t-tests were performed. t-tests were employed rather than non-parametric tests (e.g Wilcoxon test) as across tissues CCNG1 and Ki-67 both displayed normal distributions (despite being skewed in some organ subsets) and non-parametric tests may be underpowered with low sample counts such as those provided in TCGA normal tissues. We evaluated cohort-level differential expression of CCNG1 and Ki-67 in primary (n=9161) vs metastatic (n=393) across organ sites. Seeing as TCGA sources provide ample metastatic samples from skin cancer (e.g. melanoma) cases, we also performed primary vs. metastatic analysis within skin cancer specifically (n=103 and n=367, respectively). Additionally, we observed differential expression of CCNG1 and Ki-67 between settings (primary and metastasis) within the same individuals. Matched pairs of primary and metastasic tumor samples were available for 28 patients across 12 organ sites in the TCGA cohort. Fold-change was calculated by taking the ratio of primary and metastatic log2-TPM values.
Points to Consider #1: CCNG1is overexpressed throughout the TME, including adjacent non-tumorous areas.
Figure 1 shows the differential expression of CCNG1 (x-axis) vs Ki-67 (y-axis) in tumor (green) and normal (blue and red) settings. There was no significant difference in CCNG1and Ki-67 expression across organ sites in TCGA tumor and adjacent non-tumor tissue within the TME (TCGA normal). In contrast, as shown in Table 1, CCNG1 expression was significantly enhanced in TCGA tumor compared to normal tissue obtained post-mortem from patients with no tumors (GTEX normal). This is an important observation since proliferative tumor-associated microvasculature (TAM) and tumor-associated fibroblasts (TAFs) would also show enhanced expression of CCNG1 and Ki-67 in the adjacent non-tumor tissue within the TME.


Points to Consider #2: CCNG1expression is higher than that of Ki-67 in metastatic skin cancers (e.g., melanoma, basal cell carcinoma, squamous cell carcinoma).
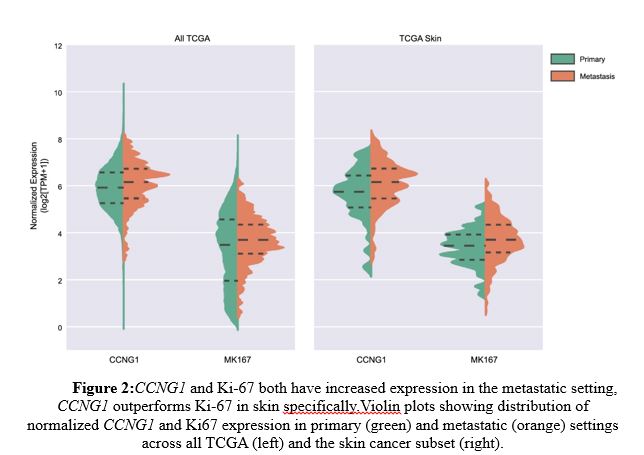
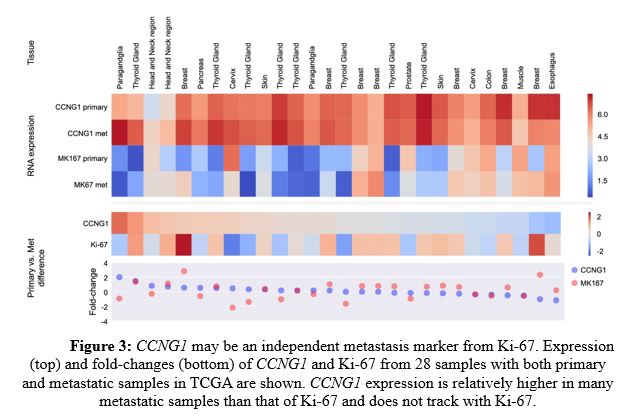
As shown in Figure 2 left panel, CCNG1 and Ki-67 both showed enhanced expression in metastatic settings (orange) compared to primary tumors (green), but CCNG1 outperformed Ki-67 specifically in metastatic skin cancer (right panel). Further, CCNG1 expression was relatively higher in many metastatic samples than that of Ki-67 and did not track with Ki-67 (Figure 3). These data indicate that CCNG1 oncogene expression may be a better indicator of metastasis than primary tumors.
Points to Consider #3: CCNG1 expression is enhanced in specific tumors by RNA sequence analysis.
As shown in Table 1, in 11 of 22 measured samples, CCNG1 RNA expression was significantly overexpressed in TCGA tumor samples compared to GTEx normal tissues, including thyroid, brain, liver, prostate, skin, testis, kidney, adrenal, stomach, pancreas and esophageal cancers.
In 4 of the 22 samples, CCNG1 RNA was under-expressed in the tumor samples, indicating that these tumors were in dormant or inactive state and as such, these tumors were not actively dividing, and may not be responsive to DeltaRex-G therapy, a retroviral based vector that integrates only in actively dividing cells [15-21].
Points to Consider #4: CCNG1 nuclear protein expression is enhanced in most tumors examined by IHC.
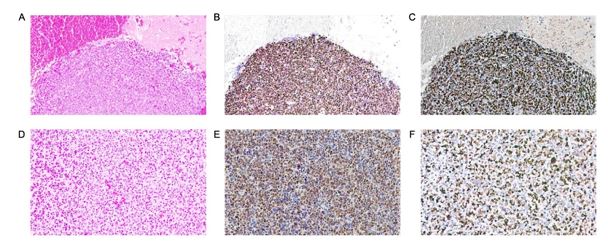
Furthermore, IHC staining revealed significant cyclin G1 overexpression in almost all tumor samples and cancer cell lines (Table 2). For example, Figure 4 shows 80% nuclear CCNG1+ and nuclear Ki-67+ tumor cells in mesenchymal chondrosarcoma metastatic to brain.
During embryological development, the CCNG1 (Cyclin G1) oncogene product normally cooperates with the TP53 (p53) family of tumor suppressor proteins in mediating genome stability: operating at multiple cell cycle checkpoints in proliferative somatic cells [27-30]. Characterization of the CCNG1 oncogene in terms of clinical oncology (dysregulated expression, viral subversion, oncogenic interactions, and stem cell survival functions) revealed Cyclin G1 also complexes with the c-Myc and Mdm2/Hdm2 oncogenes—thus identifying the pivotal Cyclin G1 / Cdk / Myc / Mdm2 / p53 Axis of cancer stem cell competence as a strategic target for broad-spectrum cancer gene therapy [3,4] and a potentially unifying theme of cancer ontogeny (Cyclin G1-complex), EMT, and metastatic progression.
Reimer et al. [31] was among the first to describe histopathological overexpression of Cyclin G1, with subcellular localization at DNA replication foci, suggesting potential diagnostic and prognostic implications of CCNG1dysregulation in both breast and prostate cancer cells in vitro, as well as cancer cells in situ from tumor specimens, using differential display polymerase chain reaction (PCR) screening [31]. Following DNA damage, Reimer et al. showed that Cyclin G1 was triggered to cluster in a discrete nuclear pattern at DNA replication foci; demonstrating subcellular co-localizing at foci containing the replication-associated proliferating cell nuclear antigen (PCNA), which physically binds to the classic CDKN1A/ p21Waf1/CIP1-tumor suppressor [32], to regulate genomic stability, cell cycle arrest, and senescence in response to DNA damage through myriad complex physical interaction(s) with TP53 (p53)[7,33].
In 2015, Jiang et al. [34] advanced the clinical understanding of CCNG1 by investigating its molecular expression and biochemical role in governing cellular proliferation and apoptosis of epithelial ovarian cancer (EOC), where CCNG1 (Cyclin G1) was found to be upregulated in EOC tumors, compared with normal ovarian tissues. In EOC, CCNG1 expression was closely correlated with differentiation grade (P = 0.009) and malignant tumor cells in ascites (P = 0.009). The Kaplan-Meier curves showed that higher expression of CCNG1 was associated with significantly shorter survival in EOC patients, while multivariate analysis suggested CCNG1 expression is an independent prognostic factor in terms of overall survival. Combined immunofluorescence and flow cytometry analysis confirmed that silencing of CCNG1 with shRNA is lethal to EOC: promoting apoptosis of ovarian cancer cells. Taken together, Jiang et al.'s findings suggest that CCNG1 may indeed serve in the prognosis of EOC patients, as well as a prospective therapeutic target.
Likewise, a molecular histological evaluation of CCNG1 expression in colorectal neoplasia, demonstrated CCNG1 overexpression in 91% of colorectal tumors studied [35]. The Cyclin G1-positive cancer patients were evenly distributed between men and women, and between tumor locations and grading. Moreover, this comparative study noted that Cyclin G1 overexpression was a more frequent event than the commonly dysregulated CCND1/PRAD1/ Cyclin D1 proto-oncogene (found in only 42% of colorectal adenocarcinomas) [35]; thereby suggesting an enhanced therapeutic potential for CCNG1/Cyclin G1 oncogene as a molecular-genetic marker and a postulated target for treating colorectal neoplasias.
In 2012, Russell et al. [36] reported that Cyclin G1 regulates the outcome of taxane-induced mitotic checkpoint arrest demonstrating that CCNG1 amplification promotes “cell survival” after paclitaxel exposure in ovarian cancer cells. Conversely, CCNG1 depletion by RNA interference enhanced paclitaxel-induced cell death via apoptosis. Further, CCNG1 amplification was associated with significantly shorter post-surgical survival in patients with ovarian cancer who had received adjuvant chemotherapy with taxanes and platinum compounds [36]. Indeed, CCNG1 overexpression per se was determined to promote epithelial-mesenchymal transition (EMT) to an aggressive “metastatic” phenotype, which is mechanistically associated with chemotherapy resistance in gastric, liver, ovarian and other cancer types [8-10, 37].
The mechanisms of oncogene addiction and cancer progression include loss of natural tumor suppression focused on CCNG1/Cyclin G1 proto-oncogene: in 2007, a series of high-throughput screens investigating the role of microRNAs in human hepatocellular carcinoma (HCC) identified miR-122 as the leading species of microRNA that was either lost or significantly down-regulated in about 70% of HCCs, and in all of the HCC cell lines [38]. Notably, these studies demonstrated CCNG1as a gene target of miR-122, due to the inverse relationship between miR-122 and CCNG1 expression seen in HCC. Loss of miR-122 gene suppression and consequential CCNG1 overexpression was associated with increased proliferation of HCC cells, disease progression, and metastasis [39], while re-expression of miR-122 was shown to inhibit both their tumorigenic properties [40] and metastatic potential [9,41]. It was confirmed that, by controlling CCNG1 expression, miR-122 impacted the stability and transcriptional activity of p53, as restored miR-122 expression reduces the metastatic invasiveness of HCC-derived cell lines [41]. Moreover, the inhibitory effect of experimentally restored miR-122 expression on CCNG1 levels increased the sensitivity of HCC cells to both sorafenib [40] and doxorubicin [41], thereby establishing a mechanistic basis for the future development of combined chemotherapy and miR-122 mimetic and/or CCNG1-based cancer therapies.
This is the first study that uses bioinformatics and a qualified IHC protocol to study a large number of tumors for CCNG1 expression compared to their analogous counterparts. As cancer treatment continues to trend toward more highly directed molecular therapies, efforts have been made to determine how to best assess which patients may benefit from specific targeted therapies. Cyclin G1 dysregulation is an important negative regulator of p53 activity, allowing for disinhibited cell cycle progression with decreasing DNA fidelity [3,4]. In the present study, we found that Cyclin G1 is overexpressed in many tumor samples; and that in metastatic tumors, Cyclin G1 expression is higher than that of the Ki-67 labeling index of cell proliferation (an important prognostic factor), providing mechanistic support for a Cyclin G1-centric view of cancer stem cell Competence, as distinguishable, in terms of aligning targets with prospective therapies, from the enzymatic and biochemical phases of proliferative cell cycle control. Together with the broad clinical efficacy of CCNG/Cyclin G1 inhibitor (DeltaRex-G) therapy, this finding supports the emerging focus on the Cyclin G1 oncogene [3,4], which should be included in the molecular-genetic profiling of tumors at diagnosis and in post-treatment tumor specimens.
DeltaRex-G is the first and, so far, only tumor-targeted retrovector encoding a CCNG1 inhibitor gene [15-22], expressed therapeutically as a gene-edited dominant-negative (dnG1), i.e., a cytotoxic Cyclin G1 pathway-blocking construct [3,4]. DeltaRex-G has been found to be of significant and durable clinical benefit in an array of advanced metastatic cancers, as salvage therapy [19-25]; therefore, the ability to screen and identify patients with specific overexpression of the commanding CCNG1 proto-oncogene can help inform clinicians as to which patients might benefit the most from CCNG1 inhibitor therapy. Additionally, finding that CCNG1 expression varied over time in some tumors suggests that genomic data from multiple biopsies or from circulating cell-free tumor DNA may be additionally informative, in relation to predicting the optimal timing and duration of DeltaRex-G and other future CCNG1 gene-based cancer therapies.
Our findings in clinical tumors—that Cyclin G1 is over expressed in tumor cells at metastatic sites when compared to primary tumors—is in agreement with clinicopathological analysis in HCC [9], wherein the pivotal role of CCNG1/Cyclin G1 expression in cancer progression is viewed as a novel prognostic biomarker and prospective therapeutic target for the clinical management of metastatic disease. One important pathway driving the epithelial-mesenchymal transition (EMT) of metastatic cancers is the stabilization of the “Snail-family” of transcription factors, described as a molecular EMT “switch” to a motile and aggressive phenotype characterized by increased invasive potential, cytoskeletal reorganization, and molecular-genetic down regulation of E-cadherin [9,42], required for maintenance of epithelial architecture. Biochemically, GSK-3β kinase phosphorylates Snail, a transcriptional repressor involved in negative regulation of E-cadherin and a marker of malignancy [43,44]. Conceptually, the Cyclin G1 Axis promotes EMT and cancer metastasis, in part, via activation of the PI3K/Akt/ GSK-3β/Snail-dependent pathway, suggesting that two parameters: Akt-activation in correlation with elevated Cyclin G1 levels may be a more powerful predictor of a poor prognosis [9,45].
This study also found CCNG1/Cyclin G1 to be overexpressed, not just in flagrant tumor cells, but within the supportive neovascular and stromal cells of the tumor microenvironment (TME) as well. The term “angiogenic switch” describes the point in tumor progression when pro-angiogenic factors begin to predominate over anti-angiogenic factors, leading to vascular proliferation in the TME, allowing the tumor to receive nutrients required to progress and to maintain its high energy demand [46,47]. Additionally, TME vasculature encourages evasion of antitumor immune surveillance, in part by impeding lymphocyte-endothelial cell interaction and upregulating immunosuppressive leukocytes [11,47]. The specific cytocidal activity of DeltaRex-G in human tumors, including proliferative tumor-associated TAMs and TAFs suggests that DeltaRex-G breaks anergy by enhancing immune cell trafficking within the TME [24]. Moreover, localized GMCSF-immune stimulation, in combination with Cyclin G1 blockade, has demonstrated promising clinical results and survival value in advanced metastatic cancers [21], while chemo-sensitization, has been experimentally demonstrated in the medical literature [40,41]. Taken together with the notable therapeutic efficacy and long-term survival milestones achieved in advanced cancers with tumor-targeted dnG1 (DeltaRex-G) [3-4,22-23,48], the pivotal and commanding role of the CCNG1 oncogene (Cyclin G1/Cdk/Myc/Mdm2/p53 Axis) of cancer stem cell Competence provides a unifying perspective for advancing new combinatorial and molecular approaches to cancer management [4,24,48-50]. Phase 2/3 studies are planned to identify patients who are likely to respond favorably and/or benefit most with DeltaRex-G gene therapy by correlating CCNG1 expression levels and treatment outcome parameters in cancer patients treated with DeltaRex-G with or without chemotherapy, targeted therapy and/or immunotherapy, revealed in the profile of oncogene expression.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
